
About us
Group leader: Tom Eirik Mollnes
Complement
Complement is part of the innate immune system protecting the host against danger from external (e.g. invading microorganisms) or internal sources (e.g. damaged tissue). Regulatory control mechanisms normally prevent local activation to become systemic. Local complement activation is necessary for fighting infections, tissue repair and regeneration. However, under various disease conditions complement is improperly activated, either locally leading to tissue damage, or systemically with risk of serious homeostatic disturbances like in sepsis and polytrauma.
Overview of the complement system
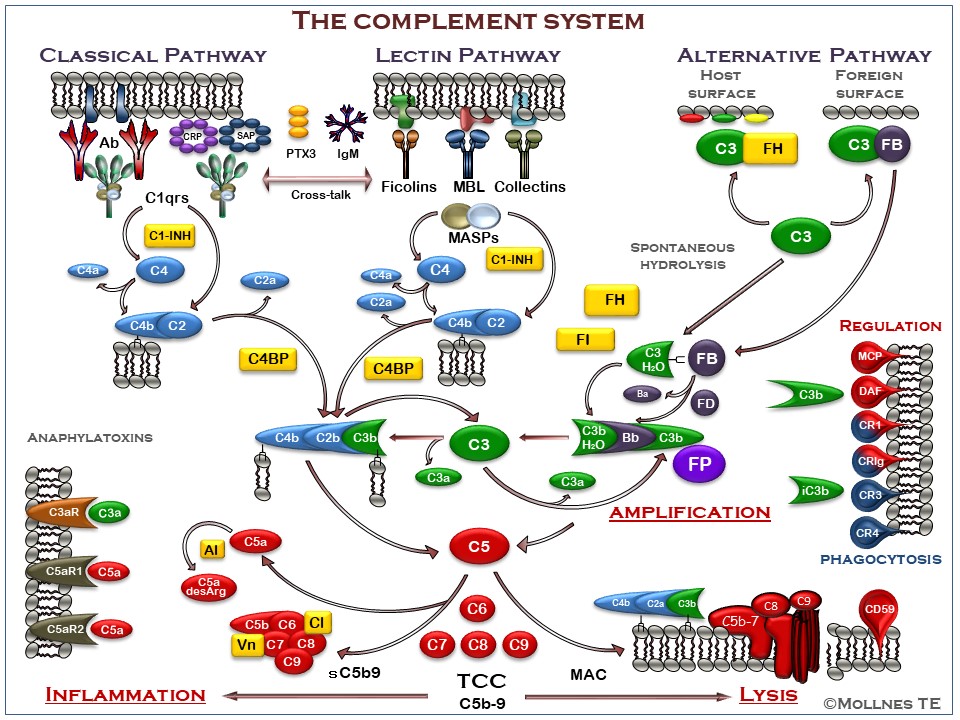
Figure legend
The complement system can be activated through three pathways (upper part of figure), all converging to the cleavage of C3 to generate C3a and C3b (middle part of figure). The classical pathway (CP) is typically activated by antibodies, but also pentraxins including C-reactive Protein (CRP), serum amyloid P component (SAP) and pentraxin 3 (PTX3) can activate C1q. The lectin pathway (LP) is activated through recognition of carbohydrates by mannose binding lectin (MBL), ficolins and collectins. Furthermore LP activation may be mediated through IgM antibodies, e.g. directed against damaged self-antigens. The alternative pathway (AP) is activated by foreign or damaged own cells, facilitated by the continuous spontaneous hydrolysis of C3. AP also has an important function in the complement system providing an amplification loop enhancing C3 activation independent of which pathway that is initially activated. This effect is mainly due to properdin (FP), the only positive regulator in the complement system, which stabilizes the C3 convertase. Activation of C3 leads to formation of a C5 convertase, cleaving C5 into C5a and C5b. The anaphylatoxins C3a and C5a bind to the receptors C3aR, C5aR1 and C5aR2, leading to downstream production of inflammatory mediators (lower left part of figure). C5b initiates the formation of the terminal C5b-9 complement complex (TCC), which either forms the membrane attack complex if inserted into a membrane (bottom part of figure). This may lead to lysis of bacteria and cells, or in sublytic doses to activation of cells. The cleavage and inactivation of C3b generates iC3b, binds to complement receptors CR3 (CD11b/CD18) and CR4 (CD11c/CD18), facilitating phagocytosis, oxidative burst and downstream inflammation (right part of figure). The complement system is tightly regulated by soluble inhibitors (marked in yellow), including C1-inhibitor (C1-INH), factor H (FH), factor I (FI), C4-binding protein (C4BP), anaphylotoxin inhibitor (AI), vitronectin (Vn) and clusterin (Cl), keeping the continuous low-grade activation in the fluid phase in check. Host cell membranes are equipped with a number of inhibitors to protect them against attack by complement (right part of figure), including membrane cofactor protein (MCP;CD46), complement receptor 1 (CR1;CD35), decay accelerating factor (DAF;CD55), controlling C4 and C3 activation, and CD59 protecting against final assembly of the C5b-9 complex.
Research goals for the NCRG
A primary research goal for the NCRG is to elucidate the role of complement as a primary inducer of the inflammatory reaction and thereby form a basis for a future therapeutic approach in complement-mediated disease processes.
For this purpose we have developed novel assays for detection and quantification of complement activation products based on monoclonal antibodies to activation dependent epitopes on a number of complement components; the most important one being the assay for the terminal C5b-9 complement complex (TCC). This antibody can detect TCC in its two different forms: the soluble sC5b-9 that can be quantified in plasma and other body fluids, and the C5b-9 inserted into membranes as the membrane attack complex (MAC) can be detected on cells and in tissues. This and several other assays are used to detect complement activation experimentally and clinically, and to evaluate the effect of various complement inhibitors in experimental models. In a human whole blood model, developed in our laboratory, where all potential inflammatory mediators are able to interact mutually, we are currently studying the effect of complement inhibition on a number of arms of the inflammatory network.
Current experimental in vitro and animal protocols, as well as clinical studies, focus on inflammatory diseases with emphasize on ischemia-reperfusion injury, transplant rejection, inflammatory response conditions like sepsis and polytrauma, and biocompatibility of medical devices. Based on the cross-talk between complement and the Toll-like receptor system, the group has put forward a hypothesis of combined inhibition of complement and CD14 as a therapeutic approach to attenuate the inflammatory reaction induced by danger signaling both from external (PAMPs) and internal (DAMPs) ligands, leading to sterile or non-sterile inflammation. A close cross-talk also exists between complement and the hemostatic systems resulting in a condition termed thromboinflammation, which is an increasingly important topic for the NCRG.
Members of the NCRG
The NCRG is lead by Professor Tom Eirik Mollnes and consists of approximately 25 members who are working at two places, the Oslo-group and the Bodø-group. Relevant information about each group member can be found in the “Members” section. Below you can find a schematic overview of the group organisation and their members.
